Definition: what is sickle-cell anaemia?
Discover our latest podcast
Also known as sickle-cell disease, it is a form of chronic and hereditary anaemia (lack of iron) and can be quite painful at times. It is a very common disease which mainly affects people with darker skin. It particularly affects people in Africa but also India and America.
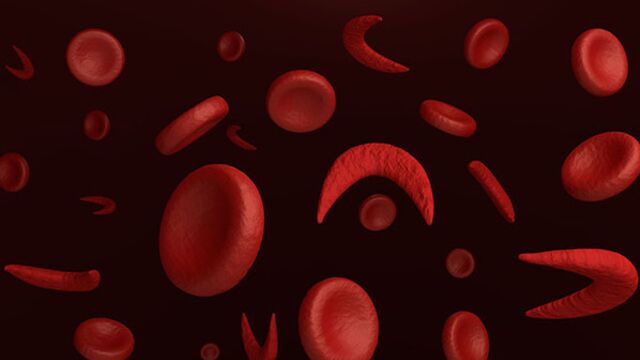
Sickle-cell anaemia is a genetic disease and is caused by the presence of an abnormal gene (allele) in the chromosome 11. This gene codes the beta chain of the haemoglobin, an essential component of red blood cells. This therefore, causes an abnormal haemoglobin called haemoglobin S or HbS which causes the red blood cells to become deformed, allowing them to take on a rigid, crescent shape.
Due to this deformation, the red blood cells are fragile and get destroyed quickly. Their life span therefore changes from around 120 days to only 20. After becoming rigid, the red blood cells also can’t pass through the smaller blood vessels and they end up blocking the blood supply to certain organs, causing circulatory problems. After a while and without care, the quick destruction of red blood cells evolves into haemolytic anaemia, which is anaemia caused by an abnormal destruction of red blood cells.
Causes: how sickle-cell anaemia is transmitted
Sickle-cell anaemia is a genetic disease, linked to a mutation of the haemoglobin gene. This condition is described as autosomal and recessive. Simply put, it is not linked to a sexual chromosome, so affects females just as much as males and it only appears when the two copies of chromosome 11 are carriers of the defective gene. Both parents must therefore be a carrier of the mutated gene. If a single parent transmits the gene, the child will also be a carrier of the faulty gene, but not suffer from the disease, however will also be a carrier.
Symptoms: how to recognise sickle-cell anaemia?
Symptoms can appear from the age of 4 months, but they widely vary over time from one individual to another. Haemolytic anaemia is the main sign but is usually accompanied by pains and an increased susceptibility to infections.
Therefore, symptoms of sickle-cell anaemia can include:
- paleness, weakness, intense fatigue, dizziness (caused by the anaemia)
- yellowing of the eyes and skin. In people with darker skin, this symptom only appears in the eyes. Darkened urine (caused by the destruction of the red blood cells)
- pains in the limbs, abdomen, belly and sometimes the bones (caused by the blockages in the smaller blood vessels)
- in children, a painful swelling of the feet and hands
- frequent infections due to an increased vulnerability. Vascular occlusion can lead to major complications such as strokes, frequent in young children who suffer from this disease.
Acute chest syndrome can also occur, due to a failure of blood circulation in the lungs which affects oxygenation. Finally, repeated occlusion can eventually cause necrosis of certain tissues.
Treatment: how to live with sickle-cell anaemia
Unfortunately, there is no safe treatment to cure sickle-cell anaemia. Current treatments essentially aim to relieve symptoms. Care involves regular monitoring and a healthy lifestyle, for which a balanced diet and good hydration are also necessary.
As well as this, it is advised to avoid factors which can trigger painful periods such as extreme temperatures or altitude. Folic acid and iron supplementation can help to stimulate the production of red blood cells and in case of increased anaemia, transfusions can be carried out. These involve transferring healthy blood to the affected person, which will help to raise the number of red blood cells.
Against the pain, painkillers can be prescribed, and a molecule called hydroxycarbamide can reduce the frequency of these attacks. However, measures are also put into place to prevent infections, through a reinforced vaccination and taking antibiotics.
In the most severe cases, a bone marrow transplant can be carried out, because this marrow contains all the stem cells necessary for blood cell production, including red blood cells.
However, it is necessary to find a compatible donor, generally a brother or a sister. If this operation is remedial it is very cumbersome and costly, and not completely devoid of risks. It is for this reason that it is only carried out in those who have a serious form of the disease.